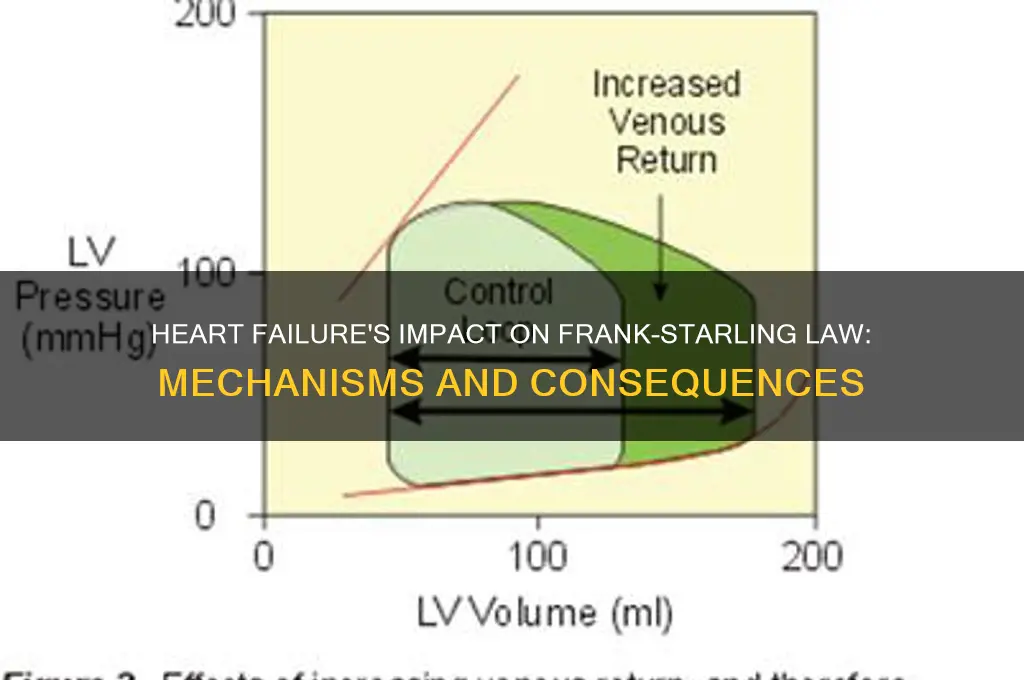
The Frank-Starling law, a fundamental principle in cardiac physiology, describes the heart's ability to automatically adjust its stroke volume in response to changes in preload (end-diastolic volume). In a healthy heart, increased venous return stretches the cardiac muscle fibers, enhancing their contractility and thereby increasing stroke volume without changes in inotropy. However, in heart failure, this mechanism becomes impaired due to structural and functional abnormalities, such as myocardial stiffness, reduced compliance, and diminished contractility. As a result, the heart fails to effectively increase stroke volume in response to elevated preload, leading to congestion, reduced cardiac output, and worsening symptoms. Understanding the dysfunction of the Frank-Starling mechanism in heart failure is crucial for diagnosing and managing this condition, as it highlights the importance of interventions targeting preload reduction and myocardial performance.
| Characteristics | Values |
|---|---|
| Mechanism | The Frank-Starling law normally allows the heart to increase stroke volume in response to increased preload (venous return). In heart failure, this mechanism becomes impaired. |
| Preload | Initially, increased preload can still augment stroke volume, but as heart failure progresses, the heart becomes less responsive to preload changes. |
| Contractility | Decreased myocardial contractility reduces the force of contraction, limiting the ability to increase stroke volume despite adequate preload. |
| Compliance | Ventricular compliance decreases, meaning the heart becomes stiffer and less able to stretch in response to increased volume. |
| Stroke Volume | Stroke volume plateaus or decreases despite increased preload due to impaired contractility and reduced compliance. |
| Ejection Fraction | Ejection fraction declines as the heart fails to effectively empty its contents during systole. |
| Compensatory Mechanisms | Early in heart failure, compensatory mechanisms like increased heart rate and neurohormonal activation (e.g., RAAS, sympathetic nervous system) attempt to maintain cardiac output, but these eventually lead to further decompensation. |
| Clinical Manifestations | Symptoms such as fatigue, dyspnea, and fluid retention arise due to the heart's inability to meet the body's demands. |
| Therapeutic Implications | Treatments focus on reducing preload (diuretics), improving contractility (inotropic agents), and modulating neurohormonal activation (ACE inhibitors, beta-blockers). |
Explore related products






What You'll Learn
- Reduced Cardiac Output - Decreased stroke volume despite increased filling pressures due to impaired myocardial contractility
- Chronic Volume Overload - Prolonged stretch leads to myocardial fibrosis, further worsening heart function
- Downward Shift in Curves - Frank-Starling relationship shifts leftward, limiting the heart's ability to respond to volume
- Neurohormonal Activation - Compensatory mechanisms like RAAS and SNS activation exacerbate heart failure progression
- Myocardial Remodeling - Structural changes in cardiomyocytes reduce compliance and distort the Starling curve
![]()
Reduced Cardiac Output - Decreased stroke volume despite increased filling pressures due to impaired myocardial contractility
In heart failure, the Frank-Starling law, which normally ensures that an increase in ventricular filling (preload) leads to a proportional increase in stroke volume, becomes impaired. This dysfunction is primarily due to reduced cardiac output resulting from decreased stroke volume despite increased filling pressures, a phenomenon driven by impaired myocardial contractility. Under healthy conditions, the myocardium stretches in response to higher preload, activating sarcomeres more effectively and enhancing systolic contraction. However, in heart failure, the myocardium loses its ability to generate sufficient force even when stretched, leading to a diminished stroke volume. This occurs because the failing heart’s myocytes are unable to effectively convert mechanical stretch into contractile force due to factors like myofilament dysfunction, energy depletion, and neurohormonal derangements.
The impaired myocardial contractility in heart failure is a key driver of this abnormality. As the heart’s intrinsic ability to contract weakens, the ventricle fails to eject a normal proportion of its end-diastolic volume, even when filling pressures are elevated. This results in a decreased stroke volume, which directly contributes to reduced cardiac output. Despite the body’s attempt to compensate by increasing preload (e.g., through fluid retention and elevated filling pressures), the failing heart cannot meet the demands of systemic circulation. This mismatch between preload and stroke volume represents a breakdown of the Frank-Starling mechanism, as the relationship between ventricular filling and contraction becomes uncoupled.
Another critical aspect is the increased filling pressures observed in heart failure. As the ventricle fails to empty adequately, blood backs up into the atria, veins, and capillaries, leading to elevated pressures in these compartments. This chronic increase in preload further stresses the myocardium, exacerbating wall tension and oxygen demand, which the failing heart is ill-equipped to handle. Over time, this leads to ventricular remodeling, including hypertrophy and dilation, which further impairs contractility and perpetuates the cycle of reduced cardiac output. Thus, the Frank-Starling law’s protective mechanism becomes a maladaptive process in heart failure.
Clinically, this dysfunction manifests as symptoms of congestion (e.g., edema, pulmonary edema) due to elevated filling pressures, alongside reduced tissue perfusion from decreased cardiac output. Treatment strategies aim to reverse or mitigate these effects by reducing preload (e.g., diuretics), enhancing contractility (e.g., inotropes), or improving overall myocardial function (e.g., beta-blockers, ACE inhibitors). Understanding the failure of the Frank-Starling mechanism in this context is crucial for managing heart failure, as it highlights the need to address both the hemodynamic consequences and the underlying myocardial dysfunction.
In summary, reduced cardiac output in heart failure arises from decreased stroke volume despite increased filling pressures, stemming from impaired myocardial contractility. This breakdown of the Frank-Starling law reflects the heart’s inability to convert preload into effective contraction, leading to a vicious cycle of congestion and inadequate perfusion. Recognizing this pathophysiology is essential for targeted therapeutic interventions to improve patient outcomes.
Driver Licensing Law: Keeping Teen Drivers Safe
You may want to see also
Explore related products






![]()
Chronic Volume Overload - Prolonged stretch leads to myocardial fibrosis, further worsening heart function
In chronic volume overload, the heart is subjected to prolonged stretch due to sustained increases in preload, often seen in conditions like valvular regurgitation or chronic kidney disease. Initially, the heart compensates by increasing its stroke volume through the Frank-Starling mechanism, which states that greater myocardial stretch leads to stronger contraction. However, this compensatory phase is not sustainable. Over time, the continuous stretching of cardiomyocytes triggers pathological remodeling. The prolonged mechanical stress activates fibroblasts and stimulates the deposition of extracellular matrix proteins, leading to myocardial fibrosis. This fibrosis replaces functional myocardial tissue with non-contractile scar tissue, which impairs the heart's ability to contract and relax efficiently.
Myocardial fibrosis in chronic volume overload disrupts the normal architecture and compliance of the myocardium. As fibrosis progresses, the ventricle becomes stiffer, reducing its ability to distend during diastole. This stiffness diminishes the heart's capacity to utilize the Frank-Starling mechanism effectively, as the relationship between preload and stroke volume becomes uncoupled. The increased stiffness also elevates diastolic pressures, leading to symptoms such as pulmonary congestion or systemic edema. Furthermore, the fibrotic tissue interferes with electrical conduction, increasing the risk of arrhythmias, which further compromise cardiac output.
The progression of myocardial fibrosis creates a vicious cycle that exacerbates heart failure. As fibrosis worsens, the heart's pump function declines, leading to further volume overload and increased wall stress. This ongoing stress perpetuates fibrotic remodeling, progressively reducing myocardial contractility. The loss of compliant myocardium means that even if preload increases, the ventricle cannot generate a proportional increase in stroke volume, effectively nullifying the Frank-Starling mechanism. This maladaptive response shifts the heart from a compensated state to decompensated heart failure, characterized by reduced ejection fraction and symptomatic deterioration.
Clinically, the consequences of chronic volume overload and myocardial fibrosis are profound. Patients often present with fatigue, dyspnea, and fluid retention due to the heart's inability to meet metabolic demands. Echocardiography typically reveals dilated chambers, impaired systolic function, and signs of diastolic dysfunction. Treatment strategies focus on reducing volume overload through diuretics, afterload reduction with vasodilators, and neurohormonal blockade to slow fibrotic progression. However, once fibrosis is established, it is largely irreversible, underscoring the importance of early intervention in volume-overloaded states to preserve the Frank-Starling mechanism and prevent heart failure progression.
In summary, chronic volume overload initiates a cascade of events that culminate in myocardial fibrosis, severely impairing cardiac function. The prolonged stretch overwhelms the compensatory mechanisms, leading to pathological remodeling that disrupts the Frank-Starling law. Fibrosis-induced stiffness and reduced contractility create a self-perpetuating cycle of deterioration, driving the transition from compensated to decompensated heart failure. Understanding this process highlights the critical need for timely management of volume overload to prevent irreversible myocardial damage and preserve heart function.
What Happened to Antitrust Laws? A Deep Dive into Their Decline
You may want to see also
Explore related products






![]()
Downward Shift in Curves - Frank-Starling relationship shifts leftward, limiting the heart's ability to respond to volume
In heart failure, the Frank-Starling mechanism, which normally allows the heart to increase stroke volume in response to increased preload (end-diastolic volume), becomes impaired. This impairment is characterized by a downward and leftward shift in the Frank-Starling curve. Under normal conditions, the curve demonstrates a steep relationship between preload and stroke volume, meaning the heart can effectively pump more blood when filled with a greater volume. However, in heart failure, this curve shifts leftward, indicating that even with the same or increased preload, the heart is unable to generate a proportional increase in stroke volume. This shift reflects a diminished cardiac reserve and reduced contractility, hallmark features of a failing heart.
The leftward shift in the Frank-Starling curve is primarily driven by myocardial dysfunction at the cellular and molecular levels. In heart failure, cardiomyocytes (heart muscle cells) undergo structural and functional changes, such as myofilament dysfunction, impaired calcium handling, and increased stiffness. These changes reduce the heart's ability to stretch and recoil efficiently during the cardiac cycle. As a result, the heart becomes less responsive to increases in preload, leading to a suboptimal stroke volume despite adequate filling pressures. This limitation exacerbates the heart's inability to meet the body's oxygen demands, perpetuating the cycle of heart failure.
Another contributing factor to the downward shift in the Frank-Starling curve is ventricular remodeling. In response to chronic pressure or volume overload, the ventricles undergo pathological changes, including hypertrophy (thickening of the heart muscle) and dilation (enlargement of the chamber). While initially compensatory, these changes ultimately impair myocardial performance. The enlarged ventricle requires more volume to achieve the same degree of wall stretch, but the myocardium's reduced contractility prevents it from effectively ejecting this increased volume. Consequently, the curve shifts leftward, reflecting the heart's diminished capacity to respond to volume loading.
The leftward shift also highlights the loss of cardiac compliance, which is the ability of the heart to distend and fill with blood. In heart failure, the stiffening of the myocardium and extracellular matrix reduces ventricular compliance, making it harder for the heart to accommodate additional volume during diastole. This reduced compliance further limits the heart's ability to utilize the Frank-Starling mechanism, as the ventricle cannot stretch adequately to activate the sarcomeres optimally. As a result, stroke volume remains low, even when preload is increased, reinforcing the downward shift in the curve.
Clinically, the downward and leftward shift in the Frank-Starling curve has significant implications for managing heart failure patients. It explains why volume overload, such as from fluid retention, can rapidly decompensate these patients, as their hearts are unable to handle the increased preload effectively. Treatment strategies often focus on reducing preload through diuretics and afterload through vasodilators, as well as improving contractility with inotropes or other therapies. Understanding this shift in the Frank-Starling relationship is crucial for optimizing care and preventing acute exacerbations of heart failure.
Trade, Anti-Legislation, and the Global Economy
You may want to see also
Explore related products






![]()
Neurohormonal Activation - Compensatory mechanisms like RAAS and SNS activation exacerbate heart failure progression
In heart failure, the Frank-Starling law, which normally ensures that cardiac output meets the body's demands by increasing stroke volume in response to higher ventricular filling (preload), becomes compromised due to impaired myocardial function. As the heart fails to pump effectively, compensatory mechanisms are activated to maintain perfusion to vital organs. Among these, neurohormonal systems, particularly the renin-angiotensin-aldosterone system (RAAS) and the sympathetic nervous system (SNS), play a central role. Initially, these systems act as protective mechanisms, but their chronic activation exacerbates heart failure progression. The RAAS is triggered by decreased renal perfusion, leading to renin release, which ultimately increases angiotensin II and aldosterone levels. Angiotensin II causes vasoconstriction to elevate blood pressure, while aldosterone promotes sodium and water retention, expanding blood volume. Similarly, the SNS increases heart rate and contractility via norepinephrine release, aiming to enhance cardiac output. However, these short-term benefits come at a cost.
Chronic activation of the RAAS leads to detrimental effects on the myocardium and vasculature. Angiotensin II not only causes systemic vasoconstriction but also promotes fibrosis, hypertrophy, and oxidative stress in the heart, further impairing its function. Aldosterone exacerbates this by increasing myocardial fibrosis and inducing ventricular remodeling, which disrupts the Frank-Starling mechanism by stiffening the ventricles and reducing their compliance. This reduces the heart's ability to stretch and fill adequately, diminishing stroke volume despite increased preload. Additionally, fluid retention due to aldosterone elevates ventricular filling pressures, imposing a greater workload on the already compromised myocardium, leading to a vicious cycle of worsening heart failure.
Simultaneously, prolonged SNS activation contributes to myocardial damage and dysfunction. Excessive norepinephrine release leads to downregulation of beta-adrenergic receptors, reducing the heart's responsiveness to sympathetic stimulation over time. This blunts the inotropic and chronotropic effects of the SNS, further limiting the heart's ability to compensate for reduced function. Moreover, catecholamine excess generates toxic oxygen radicals, causing myocyte injury and apoptosis. The increased heart rate and contractility also elevate myocardial oxygen demand, which, in the setting of coronary artery disease or reduced coronary flow, can lead to ischemia and exacerbate heart failure.
The interplay between RAAS and SNS activation creates a maladaptive feedback loop. Fluid retention from RAAS activation stimulates SNS activity to maintain blood pressure, while SNS-induced vasoconstriction reduces renal perfusion, further activating the RAAS. This cycle perpetuates volume overload, hypertension, and myocardial stress, all of which impair the Frank-Starling mechanism. The ventricles become dilated and non-compliant, unable to effectively increase stroke volume with higher preload, leading to persistent congestion and reduced cardiac output. This neurohormonal overactivity thus shifts from being compensatory to becoming a major driver of heart failure progression.
Therapeutically, targeting these neurohormonal systems is crucial in managing heart failure. Inhibitors of the RAAS, such as ACE inhibitors, ARBs, and mineralocorticoid receptor antagonists, and beta-blockers to modulate SNS activity, have been shown to improve outcomes by breaking the vicious cycle of neurohormonal activation. By reducing fibrosis, remodeling, and fluid overload, these therapies help restore ventricular compliance and partially salvage the Frank-Starling relationship, thereby slowing disease progression and improving symptoms. Understanding the role of neurohormonal activation in exacerbating heart failure underscores the importance of early intervention to disrupt these maladaptive pathways.
Barack Obama's Law License: Suspended Over What?
You may want to see also
Explore related products






![]()
Myocardial Remodeling - Structural changes in cardiomyocytes reduce compliance and distort the Starling curve
In heart failure, myocardial remodeling plays a pivotal role in altering the Frank-Starling mechanism, which normally ensures that cardiac output meets the body’s demands by adjusting stroke volume in response to changes in preload. Myocardial remodeling involves structural changes in cardiomyocytes, including hypertrophy, fibrosis, and alterations in the extracellular matrix. These changes collectively reduce the compliance of the myocardium, meaning the heart becomes less able to stretch and accommodate increased volumes of blood during diastole. As compliance decreases, the ventricle fills less efficiently, leading to suboptimal preload and a diminished ability to generate force according to the Frank-Starling law. This reduction in compliance distorts the Starling curve, shifting it downward and to the right, indicating that higher filling pressures are required to achieve the same stroke volume.
Cardiomyocyte hypertrophy, a hallmark of myocardial remodeling, contributes significantly to reduced compliance. As individual cardiomyocytes enlarge in response to increased wall stress, the myocardium becomes stiffer, impairing its ability to distend during diastolic filling. This stiffness limits the ventricle’s capacity to stretch in response to increased venous return, thereby reducing preload. Additionally, hypertrophied cardiomyocytes exhibit altered calcium handling and energy metabolism, further compromising contractile function. The combined effect of reduced compliance and impaired contractility disrupts the normal relationship between preload and stroke volume, flattening the Starling curve and diminishing the heart’s ability to respond to hemodynamic demands.
Fibrosis, another critical component of myocardial remodeling, exacerbates the reduction in compliance. Excessive deposition of collagen fibers in the extracellular matrix increases the rigidity of the myocardium, making it less distensible. Fibrotic tissue replaces functional cardiomyocytes, reducing the overall contractile mass and further impairing ventricular filling. This fibrotic remodeling not only reduces compliance but also disrupts the synchronized contraction and relaxation of the myocardium, leading to inefficient pumping. The Starling curve becomes distorted as the ventricle requires higher filling pressures to achieve even modest increases in stroke volume, reflecting the pathological adaptation of the failing heart.
The distortion of the Starling curve in heart failure has significant clinical implications. Normally, the curve demonstrates a steep relationship between preload and stroke volume, allowing the heart to adapt to changes in venous return. However, in the presence of myocardial remodeling, the curve becomes flattened, indicating a loss of cardiac reserve. This means that even small increases in preload result in minimal improvements in stroke volume, while excessive increases in filling pressure can lead to pulmonary congestion or systemic venous distension. Therapies aimed at managing heart failure often focus on reducing preload and afterload to avoid overdistension of the remodeled ventricle, highlighting the importance of understanding these structural changes and their impact on the Frank-Starling mechanism.
In summary, myocardial remodeling, characterized by cardiomyocyte hypertrophy, fibrosis, and extracellular matrix alterations, reduces myocardial compliance and distorts the Starling curve in heart failure. These structural changes impair the heart’s ability to stretch during diastole, limiting preload and compromising the normal relationship between filling pressure and stroke volume. The downward and rightward shift of the Starling curve reflects the pathological adaptation of the failing heart, where higher filling pressures are required to achieve inadequate increases in cardiac output. Understanding these mechanisms is essential for developing targeted interventions to improve ventricular function and manage the hemodynamic consequences of heart failure.
California Law: Are Dog Name Tags Mandatory for Pet Owners?
You may want to see also
Frequently asked questions
The Frank-Starling law states that the force of myocardial contraction (stroke volume) increases as the volume of blood filling the heart (preload) increases, up to a certain limit. It ensures the heart pumps out as much blood as it receives, maintaining cardiac output.
In heart failure, the Frank-Starling mechanism becomes impaired due to reduced myocardial contractility and stiffness of the heart muscle. The heart fails to increase stroke volume adequately in response to increased preload, leading to volume overload and worsening symptoms.
In advanced heart failure, the Frank-Starling law is significantly compromised. The heart’s ability to stretch and contract efficiently is lost, and further increases in preload may lead to pulmonary congestion and worsening heart function rather than improved output.
Treatments for heart failure aim to reduce preload (e.g., diuretics) and afterload (e.g., ACE inhibitors, beta-blockers) to decrease stress on the heart. Additionally, inotropes or devices like LVADs may be used to improve contractility and support the failing Frank-Starling mechanism.